Complications of LM tension: the Plummer-Vinson-Paterson-Brown-Kjelberg Syndrome (PVS)
![]()
The etiology of the PV syndrome is unclear. Does the anaemia cause the dysphagia? Does it cause the post-cricoid web? Is the dysphagia hysterical? Does the web cause the dysphagia? Is there an ectodermal defect causing the nail changes and loss of teeth. Are the labial fissures due to staph, monilia or drooling?
Answer: None of the above.
I will discuss the complications of hiatal transtraction and gastroesophageal
reflux concurrently with Plummer-Vinson syndrome (PVS) because they are identical.
The various features of PVS are merely selections from the extensive menu of
pathologic events that are directly or indirectly related to excessive LM tone
and/or the reflux it produces.
At present, PVS is widely regarded as due to an iron deficiency,(1)
perhaps complicated by a vitamin B complex deficiency.(2)
The several findings are, like the anaemia, attributed to iron deficiency or
epithelial dysplasia.
PVS is rarely seen as a full-fledged syndrome. Forme fruste occurrences
are frequent. Virtually every manifestation of PVS is also seen as an isolated
problem. The postcricoid web, once considered pathognomonic of PVS was shown
epidemiologically to occur in only 15% of females with dysphagia.(3)
The more fully expressed the syndrome, the more orad the location of the manifestations.
The classical components of the syndrome include:
Hiatus hernia
Hypochromic microcytic anemia
Sideropenia
Splenomegaly
Gastritis
Achlorhydria
Gastro-esophageal reflux
Esophagitis
Post-cricoid web
Positive vallecular sign
Loss of teeth at an early age
Glossitis
Cheilitis (rhagades, angular stomatitis, perleche)
Koilonychia
![]()
Esophagitis
The association of esophagitis with reflux is long established. The reflux
occurs as a result, not of low sphincter tone as was initially supposed, but
of transient but complete loss of lower esophageal sphincter pressure.(4)
Although reflux occurs in normal subjects,(5)
there are protective mechanisms that can prevent esophagitis in the face of
reflux. These include removal of the bulk of the refluxed material by effective
peristalsis (usually primary and provoked by swallowing) and dilution and neutralization
of the remaining acid by the more alkaline (pH 6.5 to 7.6) saliva.(6),(7)
Any impairment of salivation (e.g., by anticholinergic drugs, Sj÷rgren's
syndrome) or peristalsis (e.g., by esophagitis) favors development of esophagitis
by increasing the ACT (acid clearance time) normally 313 21 sec.(8)
A source of corrosive material is also required. This is generally the acid
(.1N HCl) of the stomach although bile acids are reputed to be equally or more
effective. Alleviating acid hypersecretion with H- antagonists tips
the balance between aggravating and relieving factors resulting in subjective
and objective improvement.(9)
Diagnosis of esophagitis
Histologically, reflux esophagitis is marked by a.) increased thickness of
the basal cell zone and b.) proximitiy of dermal papillae to the epithelial
surface. Multiple biopsies correlate poorly with the endoscopic diagnosis. Random
biopsies are only 75% positive depending somewhat on the level.
Esophagitis patients display characteristic but non-specific findings on esophageal
manometry. The amplitude of contraction is decreased, the transmission rate
is delayed a few seconds and and the duration of the contraction is shortened.(10)
In severe cases, the peristaltic wave may fail altogether.
The criteria for the diagnosis of esophagitis vary so much among authors, that
statistics as to the incidence of esophagitis with PVS, or HH for that matter,
are scarcely worth quoting at length. If the diagnosis is made with an esophagoscope,
the reported incidence tends to be higher than that reported radiologically.
The reason for this disparity is the reluctance of radiologists to make the
diagnosis unless the disease is very severe.
Radiologic diagnosis of esophagitis is not difficult. One need only have an
appreciation of the normal size of the longitudinal folds - 1 mm or less - or,
simpler still, recognize that the number of folds should be about 5-6. If 3
folds occupy the width of the contracted lumen, the mucosa is abnormal. In severe
cases only a single fold may be seen in a given projection.
Radiologists reluctance may stem from an uncertainty about the significance
of enlarged folds that is a consequence of the autoplastic theory of fold formation.
As shown in earlier, however, fold formation is a function of the circular muscle,
not the muscularis mucosae. When we rely on the number of
folds for the detection of inflammatory disease, diagnosis becomes less subjective.
In the esophagus particularly, the diagnosis becomes very easy: the patient
with well marked esophagitis will have only 2 or 3 distinct folds instead of
the normal 4-6. A convenient grading system is 5-N = grade, where N = the number
of folds. Ulcerations, stricture, wall thickening and other gross changes are
not necessary to make the diagnosis.
In the higher grades of esophagitis the primary peristaltic wave, instead of
coursing the entire length of the esophagus, dies out at the striated-smooth
muscle junction. Secondary p-waves may then partially empty the organ but are
not the clean-wiping waves which leave the esophagus empty or outlined only
by thin stripes of barium between the longitudinal folds.(11)
Esophageal folds are best seen with the patient supine. In this position the
esophagus bows, bowl-like, downward and will retain barium better than when
it forms a "hill" in the usual prone position. The same is true of esophageal
varices.
There are well known difficulties in pathologic diagnosis of esophagitis. The
material obtained at suction biopsy with the flexible scope contains only the
lamina propria. Suction biopsies contain the full mucosa but little of the submucosa.(12)
In this layer, there is no evidence of proliferative change. An increase in
the thickness of the rete layer and of the length of the mucosal "pegs" characterizes
esophagitis histologically. Such changes are normal in the distal esophagus
that is exposed to "normal" reflux levels. The presence or absence of edema
in the submucosa is never described pathologically, nor is it described in the
lamina propria for that matter. Such constraints severely limit the possibilities
for accurate pathologic diagnosis from biopsies.
The endoscopist is similarly limited because he can only see the surface of
things. The mucosal folds are obliterated in the gas-distended organ. There
is no means of judging thickness from surface appearance. Erythema, of course,
is a sign that one anticipates with inflammation, but edema, which is what the
radiologist sees decreasing the fold number, should cause a pale mucosa. It
is not generally appreciated that a barium coating magnifies depth enormously.
A crevice a few thousandths of a centimeter deep is easily seen when barium
filled because of the great density of the medium . Radiographically, these
crevasses define folds.
It has been shown that mucosal permeability is increased in esophagitis. Any resulting edema must involve a layer neither pathologist nor endoscopist can visualize. This layer can only be the submucosa.
![]()
There is some distinction made in the literature between "superficial" and "deep" esophagitis. The basis for the distinction is that "deep" esophagitis produces thickening of the esophageal wall. This seems reasonable, as in some cases of reflux the wall of the esophagus does appear grossly thickened. However, using this criterion, I found that at surgery the diagnosis of inflammatory disease involving the muscular wall of the organ was seldom verified.
There are two reasons for this, both geometrical. The esophagus shortens in conditions leading to reflux - as much as a third its length without rupture of the PEL and that much or more after rupture.Contraction converts a long narrow cylinder of esophagus to a shorter, thicker cylinder. The percent of thickening can be roughly calculated from the formula for the volume of a cylinder as this remains constant before and after contraction:
|
|
Inserting average values for the radius and length of the resting esophagus, it works out that an 8 cm shortening will increase the wall thickness 25% giving a misleading appearance of deep inflamation. |
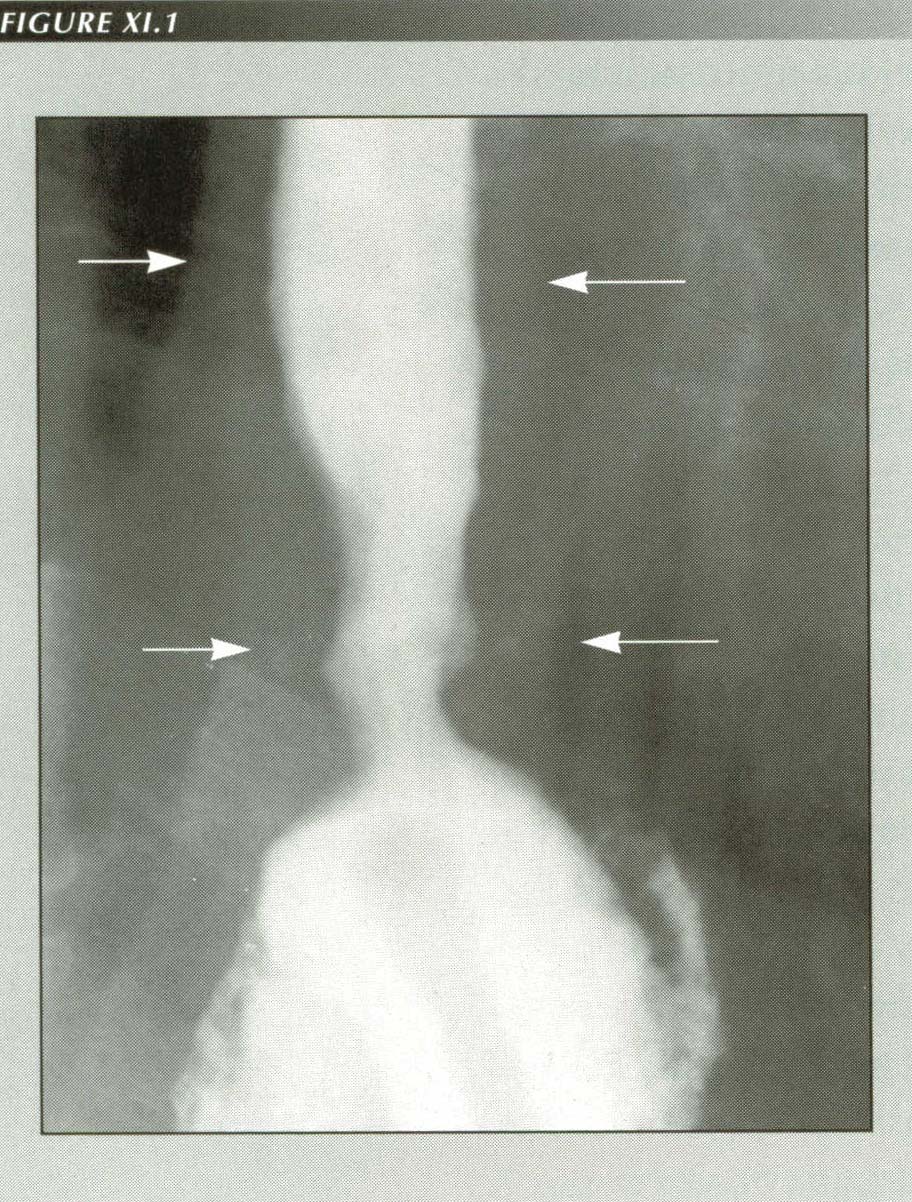
ōTerminal esophagitisö:
Schatzki believed this appearance was inflammatory, however, it occurs so frequently in patients with ruptured PELs that it seems more probable it is due to non-effacement of the sphincter. When the esophagus shortens, it becomes thicker for purely geometrical reasons. Note wall thickness (arrows)
![]()
Postcricoid webs
These were evidently first described by Kelly.(13)
Walderstrom and Kjellberg(14) documented their
association with "sideropenia." Brombart considered the finding of such webs
virtually pathognomonic of PVS,(15) however,
the more recent literature contains reports of postcricoid webs without other
manifestations of the full-blown syndrome. I have seen many postcricoid webs
in non-anemic patients ranging from a mere nick of the anterior outline of the
esophageal lumen to typical, fairly deep shelves. Like LERs they may be multiple.(16)
Also like LERs, my own cases have been associated with a HH in nearly every
instance. Seaman,(17) however, in a retrospective
study found only a 6% incidence of HH. Only 5 of his 53 patients with postcricoid
webs were classified as PVS and only 4 had gastrointestinal bleeding.
Nosher, et al.(18) reported a 5.5%
incidence of webs in 1000 consecutive cineradiographies of the throat. Of these
55 patients, dysphagia was present in only 6 and none of the patients were iron
deficient. The association of webs with dysphagia is difficult to evaluate because
"dysphagia" is frequently not defined in reports. Certainly webs can occur without
either true dysphagia or "lump in the throat" sensation. 17 of 32 webs seen
by Chisholm et al. were asymptomatic. Yet there is no doubt that a
prominent web can cause true dysphagia and that dysphagia will sometimes be
relieved by dilatation or rupture of the web.
The webs can hardly be due to anaemia per se as they also occur in
hypothyroidism and in other patients without anaemia. Yet they do occur with
increased frequency over the normal population in pernicious anaemia and with
postgastrectomy anaemia.
Postcricoid webs resemble lower esophageal rings histologically. Curiously,
the reluctance to recognize them as mucosal folds which characterizes authors
who have studied LER's is not manifested with upper esophageal webs or rings.
Entwhistle and Jacobs,(19) in 49 postcricoid
web specimens from 39 patients found that "The [histological] appearance is
essentially that of a fold of normal esophageal epithelium with some underlying
loose connective tissue." In 6 of their cases there was no evidence of inflammation
in the subepithelial tissue. A further 8 showed only a few chronic inflammatory
cells. Seven showed plasma cell and lymphocyte infiltration. Half of the 14
cases biopsied by Chisholm et al.(20)
were uncomplicated folds. The others showed similar minimal inflammatory changes.
These reports tend to show that inflammation, while it may occur secondarily,
has no part in the genesis of the web although, as with the LER, inflammation
continues to be listed as a possible cause.
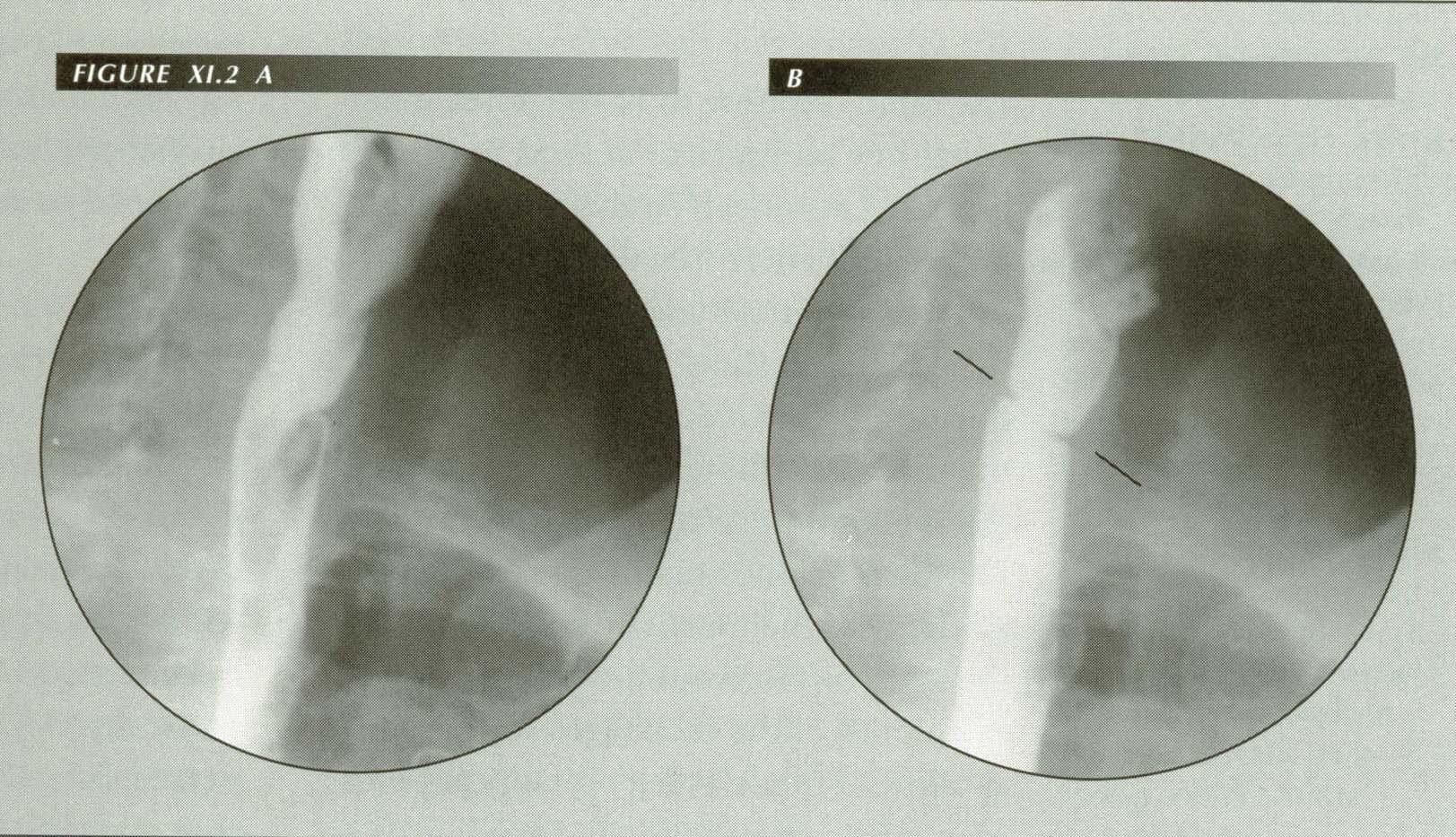
Post-cricoid ring: In many ways analogous to the LER, it is not restricted to PVS. Oddly enough, unlike LERs, there is no resistance in academia to the idea that these rings are mucosal plications. Note flow disturbance below ring.
Although these were the only 3 cases in which the state of the LM could be
determined, it is startling to learn that it was abnormal in all 3 in precisely
the area of the web. It seems almost too pat a confirmation of what fold theory
would predict. That is, if as a result of atrophy or necrosis the LM retracts
anteriorly, it could well throw up a fold. Quantitatively, however, this explanation
scarcely passes muster. Some of the webs are a centimeter in depth. It would
require over 2 cm of shortening to produce that much redundancy.
There is an association between postcricoid webs and HH. One case was reported
incidentally(21) and my impression is that they
are almost always associated, at least when the HH is over 4 cm. Smiley and
associates(22) found 19 of their series with
various hypopharyngeal "obstructions" (webs, strictures and carcinoma) also
had HH. Two had LERs as well.
The analogy of webs with the LERs is so good that there is a strong probability
that it in some fashion they have a similar cause. A web requires a source of
redundant mucosa and a reason for fixation. The following case, exceptional
in that there was no HH associated with the web, suggested that the etiology
may be similar to that of the LER.
SW CN 40987 11/12/65 Female, age 59. Fluoroscopic note: In view of the clinical
history of iron deficiency anaemia, cine films were made of several barium swallows.
These show a typical postcricoid web. The body of the esophagus was also of
interest as it appeared short constantly keeping the fundus of the stomach under
tension so that it formed a conical tent at and above the diaphragm. Because
the esophagus was under constant tension, there was free reflux spontaneously
on inspiration, during quiet respiration and with the de Carvalho test. The
sphincter stayed open for several seconds at a time.
The stomach filled to show a high subtotal resection with gastrojejunostomy.
There was very little evidence of a mucosal fold pattern in the stump.
On inquiry, the patient reported she also has a sore tongue and fissures at
the corners of her mouth both of which she herself had concluded were due to
acid reflux. She had lost her teeth at the age of 30 because of "dental caries."
The patient was reexamined a week later after 7 days iron therapy. Repeat cine
films showed that the postcricoid web had completely cleared. The esophagus
was no longer under tension and some redundancy had reappeared. The stomach
was no longer tented into the hiatus and the fundus had lost its conical appearance.
Reflux could only be elicited with the de Carvalho test.
The rapid response to Fe suggested that either low serum Fe or anaemia may
increase LM tone. If so this would result in a vicious circle.The disappearance
of the web when LM tension was relieved would tend to show that webs are also
due to mucosal redundancy. Why isn't the mucosal redundancy milked to the distal
end of the esophagus to form a LER? It is obvious that there is a need for elasticity
of the mucosa to contend with the 2 cm or more upward excursion of the mouth
of the esophagus with every swallow. If that elasticity disappears with esophagitis
or atrophy, an accordion-pleat can partially substitute. Inflammation could
also fix the mucosa to the muscularis layers.
At any rate, postcricoid webs are not pathognomonic of PVS and correlate poorly with the other features of the syndrome. There is some evidence that, like LERs, they represent mucosal redundancy occasioned by LMC, thus their association with HHs.
![]()
Hysterical dysphagia
Vinson(23) named the syndrome "hysterical dysphagia"
and, even today, this manifestation is almost the sine qua non in making
the diagnosis. As we have seen, those who have given esophageal diseases names
with etiological connotations get tagged with their mistake if they failed to
assign the correct cause. It is not surprising then to find that, in the usual
sense of the words, "hysterical dysphagia" is not dysphagia and that there is
ample evidence the symptom has an organic basis.
The "dysphagia" of PVS is sharply distinguished from true dysphagia. In the
latter, ingested food does not go down. It piles up producing substernal discomfort.
It forces the patient to stop eating. Unchanged food is regurgitated. It may
be painful or associated with weight loss.
In PVS the complaint is of "a lump in the throat." That is, the patient has
the sensation of something lodged in the throat that cannot be dislodged either
by repeated swallows, by washing it down or by regurgitating. Generally the
patient will indicate the location of the "lump" by pointing to a definite area
at the level of the thyroid cartilage. The sensation may be lateralized in patients
routinely sleeping on only one side. Occasionally he/she may report choking,
burning or coughing. Unless the patient also has a lower esophageal ring (a
common enough associated finding) or another complication, there is no real
obstruction to the passage of food.
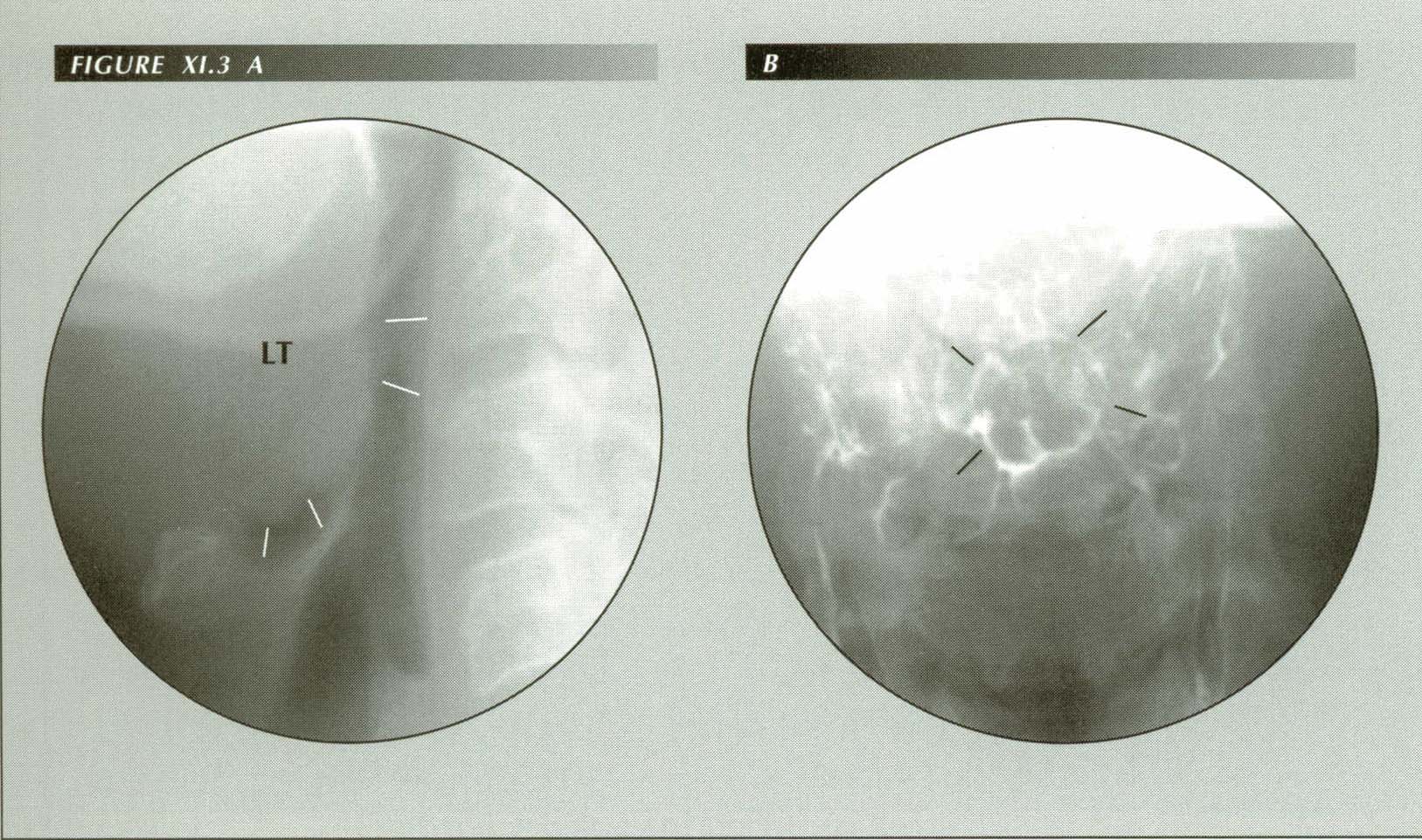
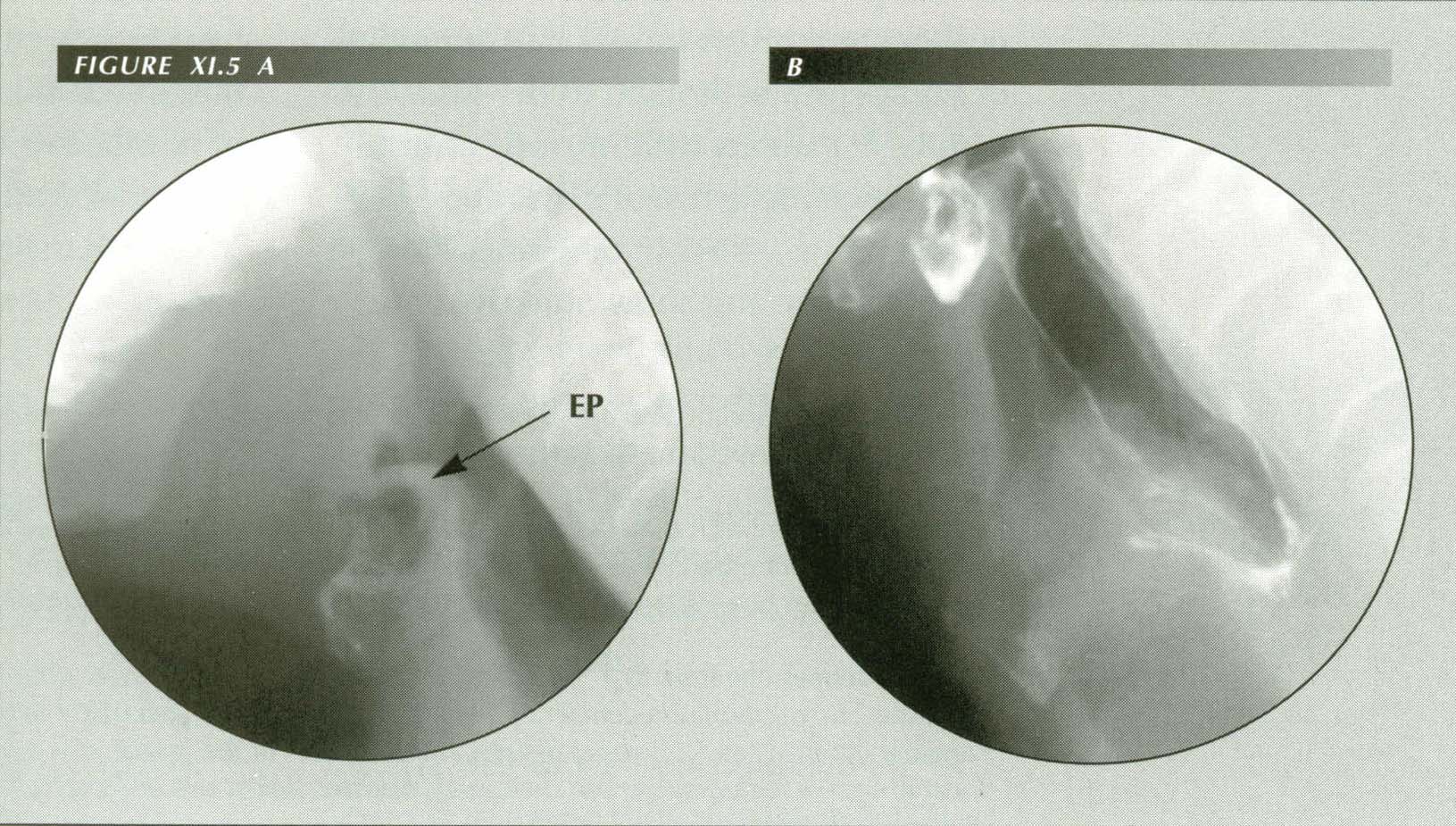
Enlargement of lingual tonsil causes globus or a ōlump in the throatö sensation. The patient states, ōIt feels like food or a pill is stuck in my throat.ö Not relieved by drinking water. Reports cheilitis but no wet spot on pillow. On fluoroscopy, there was a moderately severe esophagitis, copious reflux in response to the dC maneuver, hiatus hernia and a grade iii duodenitis. Normally, there is an air space between the tip of the epiglottis and the base of the tongue. When reflux causes edema of the epiglottis or, as is shown here, lingual tonsillitis, the air space is lost. In lateral projection (A) the epiglottis is plastered against the lingual tosil. In the frontal view (B) the contact zone appears as a nearly circular ring above the median raphe of the valleculae. This physical contact between the two structures is perceived symptomatically as a foreign object in the hypopharynx.
I found that if the patient gargles a spoonful of viscous zylocaine it produces
local anesthesia of the of the hypopharynx and eliminates the lump in the throat
sensation - clear enough proof that the symptom is not hysterical.
Although "lump in the throat" dysphagia is characteristic of PVS, it is not
limited to that syndrome. Indeed, one will see many patients with this symptom
before encountering a full-blown case of PVS. Hallewell et al.(26)
found 22 patients with HH and reflux who exhibited a lump in the throat sensation
and/or hoarseness. All found relief on antireflux therapy. Delahunty and Ardran(27)
found that of 25 patients with the globus complaint, 22 were suffering from
reflux esophagitis. The globus symptom cleared on an anti-acid regimen. They
ascribed the symptom to a motility disturbance (aperistalsis and non-peristaltic
contractions) that they were able to provoke after ingestion of "acid barium"
(pH 1.7). They also regard the motility disturbance as proof of reflux. There
were cine-radiologically demonstrated HHs in 13 patients and reflux in 10.
This work convincingly ties globus to reflux, but it does not necessarily follow
that the proximate cause of the symptom is the motility disturbance. Far more
profound disturbances of motility in "diffuse esophageal spasm" may be asymptomatic.
The most striking and unequivocal cause of the globus symptom is enlargement
of the lingual tonsil. This midline structure forms the base of Waldeyer's ring.
It is embedded in the base of the tongue directly anterior to the tip of the
epiglottis. The tip of the epiglottis is centered on the tonsil and, in the
position of rest, separated from it by an air space of several millimeters.
Because of the air space, the two are never in contact except during swallowing.
However, when the tonsil is enlarged, the two structures are in constant contact,
producing the sensation that "There is something there." that shouldn't be.
In some cases of prolonged contact, the tip of the epiglottis may create an
umbilication of the tonsil at the point of contact. In other cases more diffuse
swelling at the base of the tongue completely obliterates any air space so that
the epiglottis is plastered against the tongue base. Hypopharyngeal edema, particularly
of the epiglottis and lingual tonsil would appear to be a more direct explanation
of the globus symptom. This explains the association of the globus symptom with
reflux.
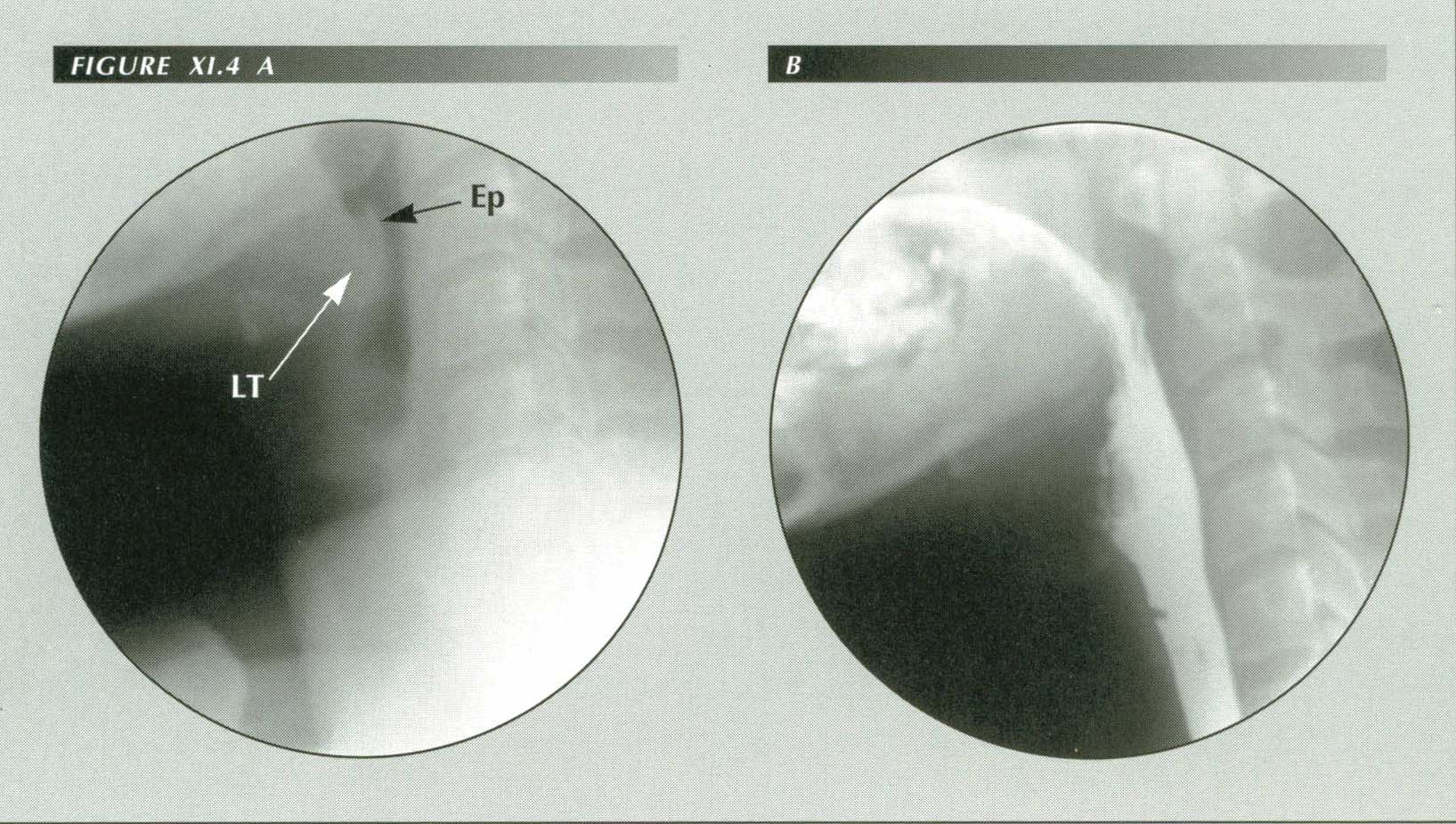
Globus due to enlarged lingual tonsil: CC: ōFeels like a peach pit caught in my throat.ö Nocturnal reflux. On fluoroscopy, grade ii reflux. Captive bolus, duodenititis, 7 cm HH, esophagitis. Normally, there is an air space between epiglottis and the lingual tonsil. An increased anterior curl of the former or enlargement of the latter brings the two in contact and this is perceived symptomatically as ōglobusö.
LT = Lingual Tonsil
Ep = Epiglottis
"Hysterical dysphagia," therefore, is neither hysterical nor dysphagia. It is hypopharyngeal consciousness due to the irritating effect of GER.
![]()
The Vallecular Sign
On radiologic examination, the valleculae usually clear of barium so cleanly
and rapidly that it may be difficult to obtain a satisfactory spot film. When
severe, hypopharyngeal inflammation is manifested radiologically by a positive
"vallecular sign"(28) -- pooling and delayed
clearing of barium from the valleculae and pyriform sinuses.
The sign is not specific. It is also seen in myasthenia gravis, senility, post
nasal drip syndrome, central or peripheral involvement of the 9th and 10th cranial
nerves, nonspecific inflammation or any condition that even slightly impairs
the hypopharyngeal swallowing mechanism.
It is frequently caused by a characteristic hypopharyngitis encountered in
patients with severe reflux. Otolaryngologists, once they have been made aware
of this cause, become quite proficient at predicting that reflux will be demonstrated
on radiologic examination. They report a "dusky reddening " of the hypopharyngeal
mucus membrane. This appearance may be passed over as normal by the laryngologist
or as an insignificant URI. Thus Cherry et al.(29)
found GER radiologically in 12 patients with unspecified "pharyngeal symptoms"
who were reported normal on pharyngeal and otolaryngological examination. Symptoms
were reproduced by perfusion of the esophagus with .1N HCl and cleared on an
antiacid regimen.
Delahunty(30) was able to demonstrate that
posterior laryngitis was caused by acid reflux, presenting 9 patients with typical
symptoms of chronic laryngitis (variable hoarseness) and laryngoscopic findings
of " . . . interarytenoid heaping of mucosa with chronic inflammation of the
posterior third of the true cords." Five also had the globus symptom. In most
cases, however, actual reflux was demonstrated. Significantly, symptoms were
relieved and the local lesion healed on antireflux therapy.
In a much larger series(31) Larrain et
al. found that 74 of 78 patients with "intrinsic" asthma showed posterior
laryngeal white plaques of varying prominence. Nearly all had pH probe-proven
reflux although most either had no symptoms of reflux or only admitted to reflux
on close questioning.
ōGlobus hystericusö: (A) The patient had reflux, esophagitis, HH, impaired p-wave and a LER. This deformity of the epiglottis can cause the globus symptom by impinging on the lingual tonsil. (B) There is no laryngeal ventricle ōfishmouthö shadow due to swelling of the false or true cords. Reflux can cause a characteristic posterior laryngitis.
Ep = Epiglottis
![]()
Pulmonary symptoms of reflux
Although not included in the classical symptom list of PVS, respiratory complications
are very frequent associated findings in GER. A history of high reflux is reliable
and far more specific for reflux than the vallecular sign. However, it is necessary
to make a specific inquiry as it is seldom volunteered. Much of the reflux occurs
at night when the patient is sleeping. Virtually pathognomonic of reflux is
a history of nocturnal laryngospasm. The patient wakes coughing, choking and
unable to get a breath. This signals high reflux with spill into the larynx.
It is a very common symptom that I have often suspected may account for some
cases of bronchiectasis. Most patients are not even aware of the reflux because
it the symptoms of laryngospasm overshadow it.
"Do you ever wake at night coughing, choking or gasping for breath?," is also
a good question to include in the routine history of patients with "hysterical"
LIT syndromes. It is astonishing that many patients with a lifelong reflux problem
never complain of heartburn, the symptom we, as physicians, associate with reflux.
They have come to believe it is a normal state of affairs! "Water brash" - the
sensation of acid rising up into the throat(32)
- occurs even during normal waking hours and correlates well with all these
radiologic findings.
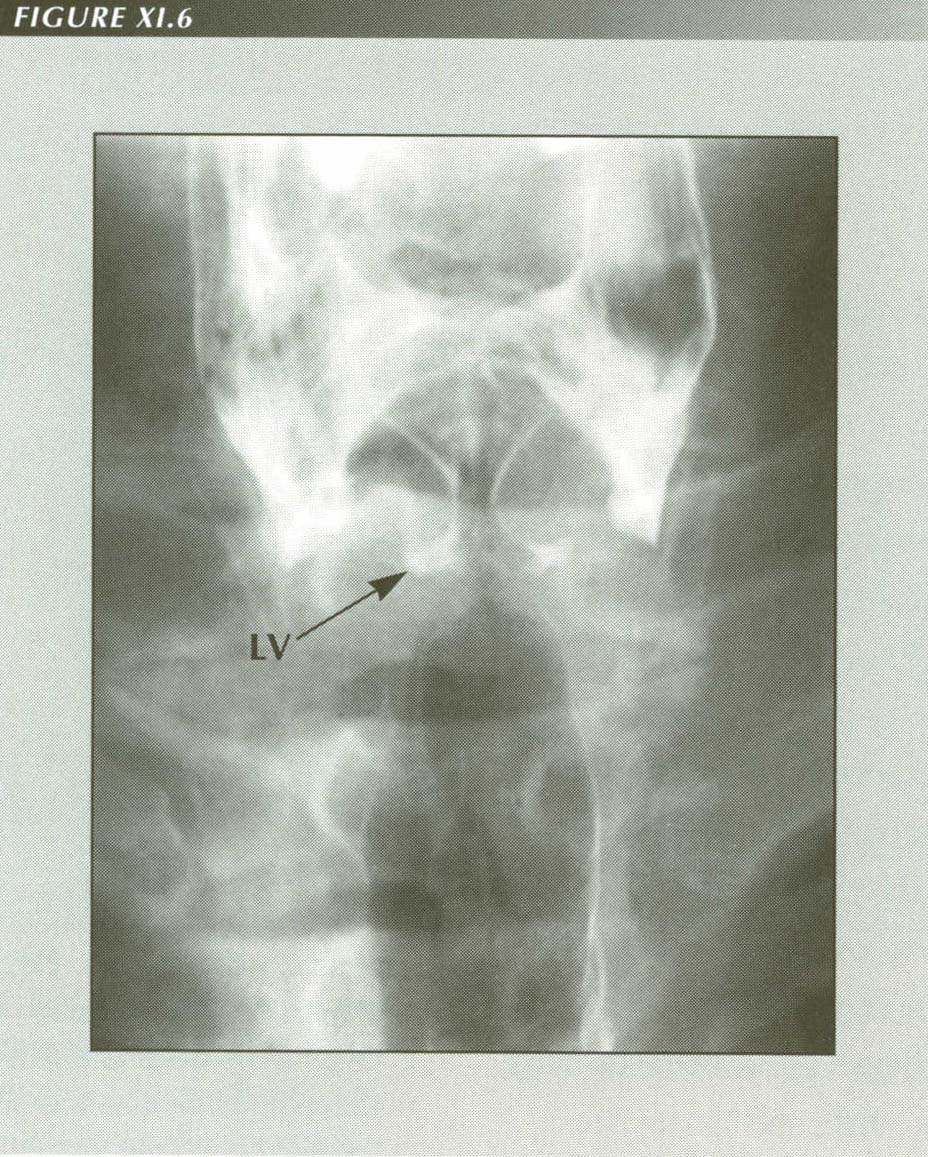
Aspirated barium defines the pyriform sinuses, true and false cords and laryngeal ventricle. Nocturnal laryngospasm is a frequent symptom of reflux. It may cause a posterior laryngitis.
LV= Laryngeal Ventricle
Tuchman, Boyle, et al.(38) showed
experimentally in cats that introduction of microvolumes (.05 ml) of .1N HCl
into the trachea increased lung resistance 4-fold by receptor mediated reflex
bronchospasm. This suggests that actual parenchymal aspiration is not necessary
for GER to alter pulmonary function.
Infants under 6 months with reflux related symptoms (apnea, choking, recurrent pneumonia, chronic cough, wheezing) had a mean duration of reflux episodes of > 6 minutes during sleep on pH monitoring.(39) Reflex bronchospasm is a possible cause of the sudden infant death syndrome.
![]()
Cheilosis
Although they are not quite synonymous, the terms cheilosis, rhagades,
angular cheilitis, perleche and lateral stomatitis are used almost
interchangeably for characteristic fissure-like erosions at the corners of the
mouth.(40) They may be crusted or denuded when
fresh, fading to slightly bluish discoloration when healed. They have long been
considered a manifestation of vitamin deficiency, particularly of riboflavin(41)
or pantothenic acid. Paterson(42) [of Paterson-Kelly]
appears to have been the first to add fissures at the corners of the mouth to
the syndrome. Their presence in PVS has suggested that the rest of the syndrome
might also be a vitamin deficiency. Goldstein(43)
states that the injection " . . . of liver extract and the administration of
vitamins have appeared to affect favorably some of the epithelial changes, especially
the cheilosis." A review of 156 cases by two oral surgeons(44)
concludes that a reduced vertical dimension of the lower half of the face was
an etiologic factor in 34% of the cases. Moniliasis is frequently incriminated
although this is a secondary infection.
The following case, in which cheilosis occurred with several signs and symptoms
due to high reflux, is one of many that suggested an explanation of the problem
that turned out to be more easily proved.
LK. CN 41221. 11/6/68 Female, age 50. History of "lump in the throat" and heartburn.
She has been seeing a dermatologist because of fissures at the corners of her
mouth. (One can still see erythematous and atrophic scars from these fissures,
which have healed under treatment.)
A preliminary film of the cervical soft tissues shows a marked anterior curl
of the epiglottis so that there is no clearance between its tip and the slightly
enlarged lingual tonsil.
Fluoroscopic note: Ingested barium passed freely through the hypopharynx and
esophagus. There were no signs of stricture or obstruction. There was pooling
in the valleculae but not in the pyriform sinuses. There were no signs of a
diverticulum.
When the patient was given a swallow or two of water in the supine position,
gross cardioesophageal reflux occurred. Some of this reached the hypopharynx
and was aspirated causing a typical episode of laryngospasm. In provoking reflux,
a sliding HH was also provoked. This was about 5 cm in length. There was a slight
LER when it was maximally provoked.
Multiple sequenced spot films show that when the HH is provoked, the slight
ring is present and the sphincter is widely patent. When the hernia reduced,
the ring disappears and the sphincter closes. They also show a slight mucosal
crinkling in the post-cricoid area.
Patients such as this suggested that, as the canthus of the mouth is dependent
while a patient is sleeping, nocturnal reflux could well cause acid burns. There
is ample evidence that patients reflux in their sleep. Drooling of acid pepsin
from the corner of the mouth seemed a reasonable etiologic speculation.
It remained a speculation for some time, but, to my routine history, I did
add the question, "Do you ever get cracks or sores at the corners of your mouth
that take a long time to heal?" There were enough affirmative answers to demonstrate
convincingly that cheilosis was not unique to the PVS.Cheilosis was a common
finding in the GE reflux population.
On receiving an affirmative reply to the above question, I routinely turned
up the lights to inspect the patient's mouth. The lesions were always bilateral
and, even if not ulcerated or crusted, they were often visible long after healing
as symmetrical, faintly bluish scars.
After a year or so, I encountered an exception to the rule of bilaterality.
The patient I was examining had involvement only on the left side of her mouth.
She was standing in the "slot" of the radiographic table, ready to be given
the first swallow of barium. I stood up to look more closely at the lesion.
Glancing down, I discovered to my delight that she had a plaster cast enveloping
her right arm and shoulder!
Other patients unable to sleep on one side or another because of casts, bursitis,
habit, etc. also proved to have lesions only on the contralateral side. As a
unilateral avitaminosis is out of the question, the explanation fits.
Although the evidence is less dramatic, on questioning, most patients with
cheilosis report finding a wet spot near their cheek on their pillow on waking
- the result of nocturnal drooling of refluxed gastric acid pepsin. Both patients
and their physicians are inclined to attribute the wet spot and cheilitis to
nocturnal drooling of saliva. However, salivation ceases during sleep(45)
as does the output of the mucus glands of the head. If further proof is required,
the patient, or in the case of children, a parent can be given an indicator
solution to test the wet spot's acidity.
This seems to dispose of avitaminosis as a cause of cheilosis per se, but does it rule out an avitaminosis causing LM tension, causing reflux, causing cheilosis? Only to this extent: It presently seems a redundant hypothesis. There are a great many patients with reflux (as the WSJ article suggests) - far more than any reasonable estimate of the number of clinical cases of nutritional deficiency.
![]()
Loss of teeth at an early age
I had always assumed that the premature loss of teeth described in PVS was
due to poor oral hygiene, lack of dental care or neglect, but the following
history, elicited from a young nurse with a severe, full-blown PVS was revealing:
"My parents spent a fortune on my teeth. I would have a lot of fillings and
then a few months later there would be another crop of cavities. Finally, the
dentist told them there was nothing he could do, that I just had "soft teeth"
and when I was 12 years old they were all extracted."
Is premature loss of teeth also due to acid reflux? The patterns of destruction,
age of onset and association with other signs and symptoms of reflux answer
this question affirmatively. The ability of acid to dissolve calcium compounds,
the known destruction of the teeth in situations with obvious exposure of the
teeth to gastric content - bulimia, cancer chemotherapy, anorexia nervosa -
point to the same conclusion. Even conditions attributed to other causes ("nursing
bottle caries") make a telling contribution to the argument.
For the past 25 years, I have routinely queried edentulous patients having
upper GI examinations as to their age when their teeth were extracted. Responses
leave no doubt that their stories resemble that of my young nurse. Remarkably,
most of them have gross reflux even though their teeth were lost decades earlier.
White(46) has documented the loss of tooth
structure associated with chronic regurgitation and vomiting. Katherine Byrne,(47)
a professional medical writer, observed in her own daughter that the enamel
of the upper incisors was first to be involved and caused these teeth to become
sensitive to hot and cold. When damaged teeth were crowned, the crowns also
became eroded.
The following account, by my dental hygienist, of the teeth of a patient with
bulimia is a graphic description of the "soft teeth" syndrome and destruction
wrought by recurrent regurgitation of acid gastric contents.
The enamel was porous - easily indented with a pick - and the teeth seemed to be shells. The buccal surfaces had been jacketed by her dentist, the lingual surfaces were eroded.(48)
![]()
Crib caries
"Crib caries"(49) are discussed here because
the mechanism of the disorder is essentially the same as that which causes loss
of the teeth in PVS. Crib caries is a rampant form of dental caries in infants
usually attributed to the custom of putting them to bed with a nursing bottle.
By the age of 2 or 3, all of the teeth may have been lost except for the lower
anterior teeth. Destruction generally begins with the upper incisors and spares
the lower teeth. Although almost any tooth may be involved, the lingual sides
are more extensively involved.(50) It is believed
that alteration of the bacterial flora by sugar in the feeding bottle promotes
caries formation. The front teeth may be spared because of their contact with
the tongue and because the high pH of the submaxillary gland saliva protects
them. It has also been theorized that the swallowing pattern of infants somehow
protects the front teeth because in infancy the tongue is thrust forward with
sucking and swallowing.
However, the same condition has been reported in infants who were breast fed.(51)
In an epidemiological study, Richardson and Cleaton-Jones (52)
rejected the "nursing bottle hypothesis" as they found that the incidence rises
with age, " . . . being far more common at five than at two, that is long after
the age of weaning . . . " Moreover, comparing the infant feeding patterns of
blacks and whites in South Africa, they found equal numbers of labial caries
among black children who did not receive fruit syrups as in white children who
did. This seems to rule out alteration of bacterial flora by excess sugar as
cause of the caries unless one makes unprovable, theory-saving assumptions (ectodermal
defect, intrinsic susceptibility, weakening of enamel by childhood diseases,
etc.
Weyers(53) classified 50 children between the
ages of 2 and 6 according to whether they had received "sugar infusions" for
prolonged periods. The statistics showed a strong inverse relationship. If anything,
drinking sugar-containing liquids from nursing bottles protected against
crib caries.(54)
I had asked the hospital dentist(55) to alert
me if he encountered an example of acid destruction of teeth. A short time later
he found such changes in a patient of 35 with a history of heartburn for years.
On examination, the same pattern of tooth destruction presented as that described
for crib caries! The front teeth were relatively spared, but the lingual surfaces
of the distal teeth were beveled down to the gingival margin. It was as though
they had been ground down obliquely with a peripheral rim of opaque enamel and
a center of semi-translucent dentine.
A striking proof of the reflux etiology of the dental abnormality was a severe
cheilosis on the left side only. The patient said that the cheilosis occurred
"every couple years" and was always on the left side - the side on which he
habitually slept when not sleeping on his stomach. The oral mucosa was discolored
(xerostomia).
On fluoroscopy, "The esophageal mucosal folds were greatly thickened, 2 of
them occupying the entire width of the esophagus. A 5 cm sliding hiatus hernia
was demonstrated with the Valsalva maneuver and gross GE reflux occurred in
response to the de Carvalho maneuver. There was no postcricoid web."
A similar case was described in a 14 year old boy by Abdulla et al.(56)
who had " . . . large numbers of chalky enamel lesions . . . of the facial and
lingual surfaces and some encircled the teeth." All of the molars had crowns
and some of the restorations had secondary caries. Xerostomia was also present
with saliva production reduced to 2 ml/hour (vs. a normal of 60 ml).
The boy had dysphagia since the age of 4. An esophageal stricture was dilated
at the age of 8. When seen in the dental clinic he had an angular cheilitis
(ascribed to B vitamin deficiency by the authors) completing the picture of
chronic reflux.(57)
Noting that patients with esophageal strictures were frequently edentulous,
Maxton et al.,(58) computed a chi-squared table
for edentulism vs. stricture for a group of 1759 patients undergoing endoscopy
at St. Thomas' Hospital and found a p < .01 that the association was due
to chance. Among a variety of explanations offered for the association (poor
nutrition from edentulism, lack of saliva causing both stricture and caries,
avoidance of solid, esophagus dilating boluses of food) they did not include
the possibility that acid reflux caused both the stricture and the
loss of teeth.
These examples, however, leave no doubt that the loss of teeth in PVS and crib
caries is not due to some ectodermal defect, vitamin deficiency or change in
eating habits but is instead due to the lytic action of hydrochloric on teeth.
The pattern of destruction is exactly what would be expected with acid reflux.
The lower anterior teeth are protected by submaxillary saliva (pH 6.5) and the
buccal surfaces of the distal teeth by parotid saliva.
We can use Occam's razor to exclude the superfluous speculations as to the
cause of crib caries. The pattern is that of acid destruction. The infants may
perform a self-administered de Carvalho maneuver by drinking - whether it be
fruit juice or mother's milk - while lying on their backs thus insuring acid
reflux by turning off the CD receptor.
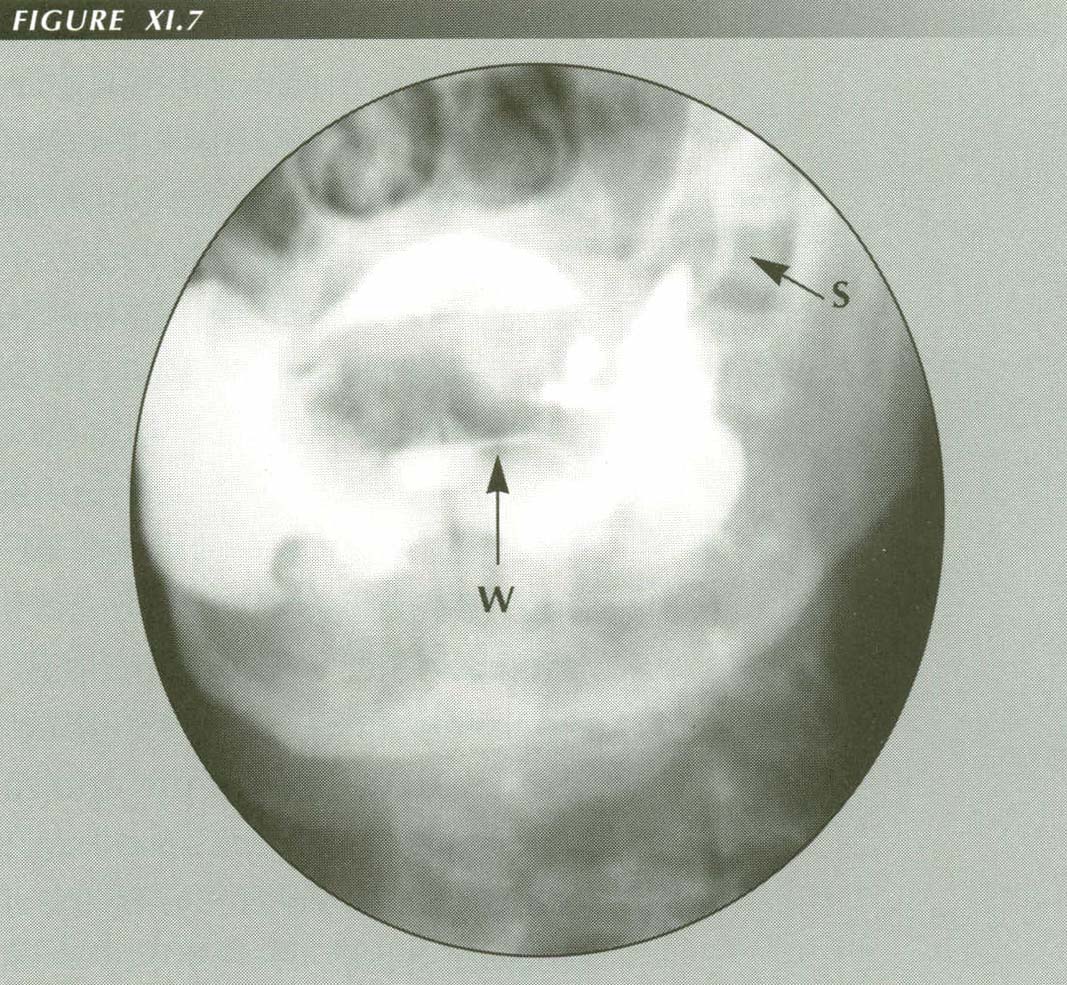
Reflux into salivary ducts: Note opacified Wharton and Stenson ducts. Apparently this was asymptomatic in this patient with neurogenic dysphagia, although if acid/pepsin were involved gland inflammation would be anticipated. Evidently this is not on the PVS palate.
W = WhartonÆs duct
S = StensonsÆs duct
![]()
Sideropenic anemia
The term "sideropenic anemia" was applied by Kjelberg who also documented the
frequency of post-cricoid webs in PVS. It is a somewhat vague term that may
be understood in several senses. "Sideropenia" - i.e., low serum iron - can
have several causes: 1.) Decreased uptake because of a) dietary deficiency or
b) impaired absorption. 2.) Increased utilization of iron. 3.) Increased iron
loss. Of these, "sideropenic" evokes the sense of dietary deficiency.
The prompt response to orally administered iron in the overwhelming majority
of cases would show that there is no defect in absorption from the gut. Percent
absorption of iron may actually be increased 300% in cases of chronic blood
loss. Malabsorption of other nutrients was never mentioned in the 183 reported
cases I reviewed.
There are local areas where the syndrome is endemic. This has suggested a lack
of iron in the soil. However, agricultural production is now international in
scope and, except in areas practicing subsistence agriculture, it is hardly
likely that a nutritional deficiency of iron could explain the incidence. The
adult iron requirement is only 1-1.5 mg/day.(59)
There is no evidence of iron deposition in the tissues in cases of PVS or other
indication of increased utilization.
Chronic gastrointestinal blood loss, however, depletes the serum iron level
by excretion of iron in the lost blood. Additional iron is then required for
hemaglobin production. The serum iron will be reduced to low values even before
the anemia is apparent. Consequently, a serum iron determination (or better
yet ferritin) is advised as a definitive test for confirmation of PVS.(60)
Chronic blood loss also explains the PVS sex ratio as menstrual blood loss
(as much as 200-500 cc per month) is additive. Superimposed on blood loss from
a HH, this is a probable cause of a severe iron loss and anaemia. In the definitive
study of blood loss in HH patients by Holt and coworkers,(61)
iron absorption and blood loss were determined with isotope methods and whole
body counting. HH patients with anemia lost an average 15 cc of blood per day,
the non anemic patients only 3 ml/day. There was no deficiency in the uptake
of iron. The anemic patientsabsorbed 39% of administered Fe vs. 8% in the non
anemic group.
The problem in sideropenic anemia, therefore, is not nutritional iron deficiency,
deficient uptake, inability to metabolize iron or iron sequestration, but chronic
blood loss. This is what ties it to hiatal transtraction and to the LMC that
causes it. HH per se, by causing chronic blood loss, can account for
the anemia of PVS. The mechanism involved will be discussed in detail in the
chapter dealing with achalasia. Here it need only be said that there is an increased
friability of the mucosa in the supradiaphragmatic portion of the fundus occasioned
by impaired venous return. This is why lesions of the fundus are more prone
to bleed.
In a series of 200 cases, Edmunds(62) found
that up to 55% of patients with "paraesophageal" hernias were anemic. Almost
the same percentage of patients with large HHs in the Mayo Clinic series (50%
of 109 cases) were anaemic.(63) On endoscopy,
a third of such patients have linear erosions on the surface of the rugae at
the level of the diaphragm producing a striped appearance on endoscopy which
has been called "watermelon stomach." Cameron and Higgins concluded that mechanical
trauma to the folds sliding through the hiatus and eroding each other was the
proximate cause. Morrissey (64)concurred, suggesting
that "The hiatus may be tight enough in some patients to cause intermittent
venous stasis." and that mechanical trauma was a factor in the friable, erythematous
appearance of the mucosa in sliding HH as well. Identical friability of the
mucosa in the herniated portion of the stomach was described by Cohen.(65)
Radiographically, fundic folds are visibly enlarged when constricted by a small
hiatus in sliding HHs as well. This is a good piece of evidence that hiatal
size can produce GE abnormalities.
Thus HH itself can produce a "sideropenic" anemia. Holt's group found that
these patients improve on iron therapy, but, because of the chronic blood loss,
they begin to go downhill when it is stopped. This creates the clinical impression
of a refractory anaemia as the blood loss is undetectable with commonly employed
guiac test.
Actually, there is no real defect in iron metabolism, no inability to absorb iron and no lack of a normal iron intake. The low serum iron, in nearly all cases, is due to chronic blood loss with consequent loss of iron and the typical hypochromic, microcytic anemia.
![]()
Splenomegaly
There is a 10% incidence of splenomegaly associated with sideropenic anemia.
The enlargement is minimal, pathogenesis is said to be unknown(66)
and there are no specific pathologic changes to suggest it is a disease sui
generis. It seems likely that the reticuloendothelial system of the spleen
merely exercises its normal function and filters abnormal microcytic RBCs from
the bloodstream.
Hiatus hernia
To complete this chain of evidence, it remains to be shown that HH is a feature
of the PVS. This has been done by Smiley, McDowell and Costello.(67)
In their series, HHs were demonstrated in nineteen of 27 patients with "pharyngo-esophageal
obstruction" (post-cricoid webs, rings, diaphragms) and other classical features
of the syndrome. The Smiley group also suspected chronic blood loss from the
HH as at least a supplementary factor in the anemia because a tendency toward
relapse suggested a continuing blood loss. However, they did not appreciate
the role of reflux in causing the buccal, lingual and pharyngeal lesions, attributing
them to " . . . faulty replacement of the foregut epithelium caused by iron-deficiency
anemia." That is, they believed it likely that an iron deficiency anemia per
se caused lingual mucosal atrophy, etc.. For this reason, and because most
HH patients do not have webs, they felt the argument for chronic blood loss
from HH as a cause of iron deficiency was flawed. The work of the Holt group,
however, clarifies this issue.
Achlorhydria
Now I must deal with the extraordinary paradox of PVS that has probably long
obscured its cause. Although I have shown that multiple symptoms and signs of
the syndrome are due to LMC causing reflux of acid pepsin, HH, etc., one characteristic
finding remains to be explained - achlorhydria! On the face of it,
this appears to destroy the entire unifying hypothesis.
Of course, one could shore it up by citing the evidence that bile acids and
other contents of the duodenum are just as corrosive as acid pepsin, but this
would mean that we would have to postulate that there was pyloric incompetence
or reverse peristalsis in the duodenum. There seems no reason to make that assumption.
In actuality, no further assumptions are required. Achlorhydria is one of the consequences of the anemia. The mechanism is as follows:
1.) LMC causes reflux and hiatal transtraction
2.) One or both of these result in chronic blood loss.
3.) The resulting anemia per se causes a superficial gastritis that, if untreated, progresses to atrophy of the gastric mucosa.
4.) Atrophy of the gastric mucosa causes achlorhydria.
The atrophic mucosa and achlorhydria of pernicious anemia are classical. Perhaps
less well known are the changes that are seen in "iron deficiency" anemia. Davidson
and Markson(68) studied 42 patients and 39 age-matched
controls with gastric biopsy. They found that the gastric mucosa was abnormal
in three quarters of the patients with iron-deficiency anemia. When histamine-fast
achlorhydria was also present, the mucosa was abnormal in 95% of cases. The
abnormalities ranged from chronic superficial gastritis to atrophic gastritis
to gastric atrophy. In most cases the lesion was an atrophic gastritis.
The incidence of achlorhydria was 48% in the anemia group vs. 13% in the controls
(who were other hospital patients without anemia). In 5 patients in whom the
hemoglobin level returned to normal after iron treatment, free Hcl reappeared
in the gastric secretions of the two with superficial gastritis but not in the
2 with atrophic gastritis. The 5th patient had free HCl prior to treatment despite
a severe anemia and superficial gastritis.
Leonard(69) found that of 47 cases of hypochromic
anemia in military inductees, 13 had achlorhydria of which 6 were reversible
on iron therapy. The frequency of achlorhydria was higher in the groups with
the lower hemoglobin levels. In a series of 50 patients with hypochromic anemia,
Badenoch et al.(70) also found that
43 (86%) had abnormal gastric mucosa. They noted that "There was a good correlation
between the severity of the mucosal changes and the incidence of histamine-fast
achlorhydria."
Two circumstances influenced Davidson and Markson(71) to resolve the question of which came first - gastritis or achlorhydria - in favor of the gastritis.
Their experience and the experience of others that treatment of the anemia restored free HCl.
In the milder superficial gastritis, 50% of the patients retained the ability
to produce gastric HCl.
The alternative hypothesis is that a primary achlorhydria could lead to faulty
absorption of iron and so cause an anemia. However, in PVS the blood loss is
the cause of the anemia so there is no point in postulating an idiopathic achlorhydria
as well.
Badenoch et al. also concluded the achlorhydria was secondary to the
gastritis and that gastric mucosal changes, like koilonychia and "angular stomatitis"
are a result rather than a cause of iron deficiency.
While concurring in this order of precedence, I would have to demur that, since atrophic gastric mucosa also occurs in pernicious anemia, it is more reasonable to conclude that the mucosa atrophies because of the anemia than because of lack of iron. As has been noted, the cheilosis is secondary to reflux.
![]()
Koilonychia
It is not quite as clear that the koilonychia is due to anaemia rather than
low iron levels in the blood serum. Like the anemia, it clears when massive
doses of iron are administered(72). If nail
changes were seen in pernicious anaemia with normal iron levels, it would suggest
that iron was not the cause of the altered nail growth. An the other hand, nail
changes are common in conditions with low O2 saturation levels -
cyanotic heart disease and chronic pulmonary disease - suggesting that anemia
per se is the cause of the nail disorder.
SUMMARY
I have presented the evidence that the LM of the esophagus causes each of the
features of PVS. It does this by producing both hiatal transtraction and reflux.
The former leads to chronic blood loss, anemia, sideropenia, gastric atrophy
and achlorhydria. Intractable reflux leads to esophagitis, hypopharyngitis,
laryngitis, glossitis, dental destruction and cheilosis, i.e., chemical trauma
to the esophagus, pharynx, larynx, tongue, teeth and skin. LERs and, possibly
postcricoid webs, are other manifestations of the LMC that causes the reflux.
"Hysterical dysphagia" is neither hysterical nor dysphagia but is due to chemical
hypopharyngitis and/or lingual tonsillitis. The high incidence of cancer of
the hypopharynx and esophagus is referable to the chemical insult of long duration.
Anemia, whether sideropenic or pernicious, leads to atrophy of the gastric
mucosa and, eventually, to achlorhydria. Other pathologic entities attributable
to reflux are identified. These include posterior laryngitis, a simulated "bronchial
asthma" and pulmonary fibrosis with some cases of bronchiectasis suspect.
It is clear that some agent or agents can cause relentless hyperfunction of the LM of the esophagus leading, in this syndrome at least, to life-threatening consequences. The fundamental problem for esophageal research is to discover the nature of these agents and means of counteracting them.
![]()
References
Last Updated July 30, 2007by David PJ Stiennon
1. Fortunately, Sir Arthur Hurst's conjecture that the syndrome could be due "...to involvement of Auerbach's plexus...thereby interfering with the normal neuromuscular mechanism, in a way similar to that found in achalasia of the cardia." did not enjoy the same success in muddying the waters at the upper end of the esophagus as that achieved by his achalasia hypothesis at the lower end.
2. 1. Roth, James L. A., Reflex esophagitis: classification In: Bockus Gastroenterology, Fourth Edition, Vol. 2, Ed. Berk, J. Edward, W.B. Saunders Company, Philadelphia, 1985.
3. 2. Elwood, P.C., Jacobs, A., Pitman, R.G. and Entwhistle, C.C., Epidemiology of the Paterson-Kelly syndrome. Lancet 2: 716-20, 1964.
4. 3. Hauser, R., Dodds, Wylie J., Patel, G.K., et al., Mechanism of GER in patients with reflux esophagitis. (Abstract) Gastroenterology 75: 1153, 1979.
5. 4. Dodds, W.J., Dent, J., Hogan, W.J., et al., Mechanisms of gastroesophageal reflux. NEJM 307: 1547-52, 1982.
6. 5. Helm, J.F., Dodds, Wylie J., Hogan, W.J., et al., Flow and acid neutralization capacity of human saliva. Gastroenterology 80:1181, 1980.
7. 6. Helm, James F., Dodds, Wylie J., Pelc, Lorie R., et al., Effects of esophageal emptying and saliva on acid clearance from the esophagus. NEJM 310:284-8, 1984.
8. 7. Helm, J.F., Riedel, D.R., Dodds, Wylie J., et. al., Determinants of esophageal acid clearance in normal subjects. (Abstract) Gastroenterology 80:1181, 1980.
9. 8. Wesdorp, E., Bartelsman, J. Pape, K., et al., Oral cimetidine in reflux esophagitis: a double blind study. Gastroenterology 74:821-4, 1978.
10. 9. Kahrilhas, P.J., Dodds, W.J., Hogan, W.J., Kern, M., Arndorfer, R.C. and Reece, A., Esophageal peristaltic dusfunction in peptic esophagitis. Gastroenterology 91 897-904, 1986.
11. 10. Ismail-Beigi, Farhad and Pope, Charles E., Distribution of histological changes of gastroesophageal reflux in the distal esophagus of man. Gastroenterology 66:1109-13, 1974.
12. 11. Goldman, Harvey and Antonioli, Donald A., Mucosal biopsy of the esophagus, stomach, and proximal duodenum. Human Path. 13 423-32, 1981.
13. 12. Kelly, A.B., Spasm at entrance of esophagus. Proc. Royal Soc. Med., Lond. Sect. Laryngol. 34: 285-9, 1918.
14. 13. Walderstrom, J. and Kjellberg, S.R., The roentgenologic diagnosis of sideropenic dysphagia, Acta Radiologica 20:618, 1939.
15. 14. Brombart, Marcel, Clinical radiology of the esophagus, John Wright & Sons, Bristol, 1961.
16. 15. Stiennon, O. Arthur, The anatomic basis for the lower esophageal contraction ring. Amer. J. of Roentgenol. Rad. Ther. & Nuclear Med. 90:811-22, 1963.
17. 16. Seaman, William B., Significance of webs in the hypopharynx and upper esophagus. Radiology 89:32-38, 1967.
18. 17. Nosher, J.L., Campbell, W.L. and Seaman, W.B., The clinical significance of esophageal and hypopharyngeal webs. Radiology 117:45-7, 1975.
19. 18. Entwhistle, C.C. and Jacobs, A., Histological findings in the Paterson-Kelly syndrome. J. Clin. Path. 18:408-13, 1965.
20. 19. Chisholm, Morag, Ardran, G.M., Callander, Sheila T. and Wright, Ralph, A followup study of patients with post-cricoid webs. Quart. J. Med (New series) 159:409-420, 1971.
21. 20. Stiennon, 1963, op cit.
22. 21. Smiley, T.B., McDowell, R.F.C. and Costello, W.T., Sideropenic dysphagia and hiatus hernia. Lancet 2:7-11, 1963.
23. 22. Vinson, P., Hysterical dysphagia, Minnesota Med. 5:107, 1922.
24. 23. Schatzki, Richard, Globus hystericus. NEJM 270: 675, 1964.
25. 24. Radiological findings in globus hystericus. Brit. J. Radiol. 39:583-6, 1966.
26. 25. Hallewell, John D. and Cole, T. Boyce, Isolated head and neck symptoms due to hiatus hernia. Arc. Otolaryng. 92:499-501, 1970.
27. 26. Delahunty, D.J. and Ardran, G.M., Globus hystericus -- a manifestation of reflux esophagitis? Ann. Allergy 29:1049-54, 1971.
28. 27. Arndt, S. and Wolfe, A., Vallecular sign, Amer. J. of Roentgenol., Rad. Therapy & Nuclear Med. 57:435
29. 28. Cherry, Jerrie, Siegel, Charles I., Margulies, Stanley I and Donner, Martin, Pharyngeal localization of symptoms of esophageal reflux. 912-5, 19
30. 29. Delahunty, J.E., Acid laryngitis. J. Laryngol. & Otology 86:335-42, 1972.
31. 30. Larrain, A., Lira, E., Otero, M. and Pope, C.E. II, Posterior laryngitis -- a useful marker of esophageal reflux. Gastroenterology 90:1986.
32. Wisconsin definition. It seems to be a folk medicine term which elsewhere may mean hypersalivation. You have to ask.
33. 31. Babb, Richard, Notarangelo, Joseph and Smith, Vernon M., Wheezing: a clue to gastroesophageal reflux. Am. J. Gastroenterol. 53-54:230-33, 1970.
34. 32. Klotz, S.D. and Moeller, R.K., Hiatal hernia and intractable bronchial asthma. Ann. of Allergy 29:325-8, 1971.
35. 33. Pearson, J.E.G. and Wilson, R.S.E., Diffuse pulmonary fibrosis and hiatus hernia. Thorax 26:300-5, 1971.
36. 34. Larrain, A, Carrasco, J., Galleguillos, C.E. and Pope, C.E., Reflux treatment improves lung function in patients with intrinsic asthma. (Abstract) Gastroenterology 90:1204, 1986.
37. Contrary opinion has it that, because both intrinsic and extrinsic asthma sufferers have a high incidence of reflux, the bronchodilators used in treatment of asthma may cause reflux by relaxing the LES. However, this hypothesis does not explain relief of asthmatic symptoms by antireflux treatment.
38. 35.Tuchman, David N., Boyle, John T., Pack, Allan I., et al., Comparison of airway responses following tracheal or esophageal acidification in the cat. Gastroenterology 87:872-81, 1984.
39. 36. Jolley, Stephen G., Herbst, John J., Johnson, Dale G., Matlak, Michael E. and Book, Linda S., Esophageal Ph monitoring during sleep identifies children with respiratory symptoms from gastroesophageal reflux. Gastroenterology 80:1501-6, 1980.
40. Dorland defines cheilosis as 1 "A condition marked by fissuring...of the lips and angles of the mouth. It is characteristic of riboflavin deficiency." Lindau's International Dictionary of Medicine and Biology defines "angular cheilitis" as "...chronic fissures at the corners of the mouth. It is a feature of a variety of general and local disorders including certain deficiency diseases, infections and problems which result in salivary spillage at the corners of the mouth."
41. 37. Meulengracht, E., and Bichel, J., Riboflavin avitaminosis und das Plummer-Vinson syndrome, Klin. Wochenschr. 20:831, 1941
42. 38. Paterson, D.R., J. Laryng. Rhin. & Otol. 34:289, 1919.
43. 39. Goldstein, F., In Gastroenterology, Vol 1, Ed. Bockus, Henry L., W.B.Saunders Company, Philadelphia, 1963.
44. 40. Konstantinidis, Antonis and Hatziotis, John H., Angular cheilosis: an analysis of 156 cases. J. Oral Med. 39:199-205, 1984.
45. 41. Schneyer, L.H., Pigman, W., Hanahan, L. et al., Rate of flow of human parotid, sublingual and submaxillary secretions during sleep. J. Dent. Res. 35:109, 1956.
46. 42. White, D.K., Loss of tooth structure associated with chronic regurgitation and vomiting, J. Am. Dent. Assoc. 110:833-35, 1978.
47. 43. Byrne, Katherine, A parent's Guide to Anorexia and Bulimia, Shocken Books, New York, 1987.
48. 44. Kathy Burger, RDH, Madison, WI, personal communication.
49. 45. Nursing bottle caries, Shelton, P.G., Berkowitz, R.J. and Forrester, D.J., Pediatrics 59:777, 1977.
50. 46. Brams, Michael and Maloney, Joseph. "Nursing bottle caries" in breast fed children. J. Pediatr. 103 415-6, 1983.
51. 47. Brams and Maloney, op cit..
52. 48. Richardson, Barbara D. and Cleaton-Jones, Peter E., Letter to the editor, ASDC J. Dent. Child. 50:72, 1983.
53. 49. Weyers, H., Untersuchungsbefunde bei "Nursing-bottle-caries." Dtsch. Zahnõrztl. Z. 38:722-6, 1983.
54. Several of the patients illustrated in Weyers' paper appear to have angular stomatitis. Another has a "geographic tongue".
55. Bruce Fischer, D.D., Tomah, Wisconsin Veterans Administration Hospital.
56. 50. Abdullah, Fawzia, Derkson, Gary and Krasse, Bo, Rampant caries due to an uncommon medical problem: report of a case. Can. Dent. Assoc. J. 50:711-3, 1984.
57. The authors, however, attributed the caries to alteration of oral bacterial flora incident to changes in eating habits due to the necessity of selecting easily swallowed foods.
58. 51. Maxton, D,G., Ainley, C.C., Grainger, S.L., et al., Teeth and benign oesophageal stricture. Gut 28:61-3, 1987.
59. 52. Wintrobe, M.M. and Lee, G.R., Iron deficiency anaemia and other hypochromic microcytic anemias. Harrison's Principles of Internal Medicine, Sixth Edition, Eds: Wintrobe, Maxwell M., Thorn, George W., Adams, Raymond D., Bennett, Ivan L., Braunwald, Eugene, Isselbacher, Kurt J. and Petersdorf, Robert G., McGraw Hill Book Company, New York, 1974.
60. The depletion is so marked the diagnosis can be made on inspection of the supernatant serum in the sedimentation tube.
61. 53. Holt, J.M., Mayet, F.G.H., Warner, G.T., Callender, S.T. and Gunning, A.J., Iron absorption and blood loss in patients with hiatus hernia. Brit. Med. Jr. 3:22-5, 1968.
62. 54. Edmunds, V., Hiatus hernia, a clinical study of 200 cases. Quart. J. Met 26:445-65, 1957.
63. 55. Cameron, Alan J. and Higgins, John A., Linear gastric erosion: a lesion associated with large diaphragmatic hernia and chronic blood loss. Gastroenterology 91:338-42, 1986.
64. 56. Morrissey, John F., Editorial: You see what you look for. Gastroenterology 91:481-2, 1986.
65. 57. Cohen, Bernard, In: The esophagogastric junction. Proceedings of a symposium held in Las Croabas, Puerto Rico, Eds. Katz, David and Hoffman, Fredrick. Excerpta Medica, Princeton, 1971.
66. 58. Wintrobe, MM, Lee, G. Richard, Boggs, Dane R., et al., Clinical hematology. 7th Edition, Lea & Ferbiger, Philadelphia, 199%%%.
67. 59. Smiley, T.B., McDowell, R.F.C. and Costello, W.T., op cit.
68. 60. Davidson, W.M.B. and Markson, J.L., The gastric mucosa in iron deficiency anemia. Lancet 2:639-43, 1955.
69. 61. Leonard, B.J., Hypochromic anemia in RAF recruits. Lancet 1:899-902, 1954.
70. 62. Badenoch, J., Evans, J.R. and Richards, W.C.D.m The stomach in hypochromic anemia. Brit. J. Hemat., 3:175-85, 1957.
71. 63. Davidson, W.B.M and Markson, J.L., op cit..
72. 64. Hallen, Lars, Acta Medica Scand. Suppl. 90:398-405, 1938.